
Épendymome
Cet article est une ébauche concernant la médecine et la neurologie.
Vous pouvez partager vos connaissances en l’améliorant (comment ?) selon les recommandations des projets correspondants.

| Spécialité | Oncologie |
|---|
| CIM-10 | C71 |
|---|---|
| CIM-9 | 191.9, 225.0, 237.5 |
| ICD-O | M9391/3-9394/1 |
| DiseasesDB | 29452 |
| eMedicine | 277621 |
| MeSH | D004806 |
L'épendymome est une tumeur du système nerveux central de type kystique. Dans la majorité des cas il s'agit d'une tumeur bénigne (grade I ou II), mais parfois elle peut être maligne (cancéreuse) et être alors de grade III (anaplasique). Cette maladie fait partie de la famille des gliomes.
Généralités
[modifier | modifier le code]Les épendymomes (EP) sont une famille de tumeurs cérébrales prenant naissance dans les cellules gliales qui tapissent les points de passages du liquide céphalo-rachidien [1]. Il s’agit de tumeurs rares chez l’enfant et représentent en France 6.5% des tumeurs du système nerveux central. Ils regroupent différents types de tumeurs de présentation clinique et de pronostic différent. Selon le Registre central des tumeurs du cerveau des États-Unis, leur fréquence est plus élevée chez les enfants, soit 5,2 %, tandis que chez les adultes, elle est d'environ 4 %[2]. Ces tumeurs sont plus souvent retrouvées chez les femmes que chez les hommes (1,3:1). La localisation de la tumeur dépend largement de l'âge du patient, ainsi environ 90 % des épendymomes pédiatriques sont retrouvés dans le cerveau tandis que 65 % des tumeurs adultes surviennent dans la moelle épinière [3].
Les épendymomes sont des tumeurs très hétérogènes (on dénombre à ce jour plus de 10 types tumoraux différents). Leur classification actuelle repose sur des critères histologiques et moléculaires afin de capturer au mieux cette diversité car cela permet de prendre en charge les patients de façon plus ciblée et personnalisée. Le traitement des épendymomes repose sur de la radiothérapie associée à une résection chirurgicale lorsque cela est possible. La chimiothérapie est quant à elle réservée aux cas ne pouvant pas être réséqués sans atteinte majeure du cerveau [3]. [4].
Classification de l’OMS
[modifier | modifier le code]Plus précisément, les EP sont classés en fonction d'une combinaison de caractéristiques histopathologiques et moléculaires, ainsi que de leur site anatomique. On distingue 3 localisations différente: supratentorielle, fosse postérieure (PF) et spinale. Ces groupes sont ensuite divisés en fonction de leurs altérations moléculaires, qui comprennent par exemple des fusions oncogéniques pour les épendymomes supratentoriels (ZFTA/RELA et YAP1/MAMLD1), des pertes de méthylations d’histone pour les EP PF-A ; ou encore des amplifications géniques (MYCN) pour les EP de la moelle épinière [5]. La dernière révision de la classification OMS parue en 2021 reconnaît pour chacun de ces types tumoraux un profil épigénétique particulier, qui peut être déterminé par l’étude du méthylome de la tumeur. Cette technologie est en pleine expansion dans le domaine de la neuropathologie tumorale. Enfin, l’OMS s’accorde à classer les tumeurs en fonctions de différents grade histologiques allant du grade 1 au grade 3. Plus le grade est faible, plus les cellules tumorales ressemblent à des cellules normales et moins elles prolifèrent ; plus le grade est élevé, plus les cellules sont indifférenciées et présentent des atypies marquées. Ces tumeurs sont alors associées à une prolifération élevée et un risque d’échappement thérapeutique.
Symptomes
[modifier | modifier le code]Les symptômes de l’épendymome vont varier en fonction de la localisation et de la taille de la tumeur.
Ependymome cérébraux :
Une compression par la tumeur au niveau du cerveau peut provoquer de nombreux symptômes tels que des troubles de la vision, des maux de tête réguliers, ainsi que des problèmes ORL comme des troubles de la vision et/ou des pertes d’équilibres [6].
Ependymome intramédullaire :
Les possibilités de compression de la moelle épinière sont diverses :
- Au niveau des cervicales avec des problèmes de sensibilité, des fourmillements ainsi que des problèmes moteurs et de maîtrise des sphincters.
- Au niveau des lombaires, qui sont les compressions les plus fréquentes avec des troubles urinaires, sensitifs et/ou des faiblesses musculaires pouvant entraîner une paralysie des membres [6].
Épendymomes supratentoriels
[modifier | modifier le code]Les épendymomes supratentoriels (SE) représentent entre 40% et 60% des tumeurs intracrâniennes. Localisées au niveau supratentoriel, ces tumeurs apparaissent comme des grosseurs peu homogènes, présentant des zones kystiques, des calcifications mais également des zones hémorragiques et nécrosées.
Aspect radiologique à l’IRM en séquence T1 après injection de Gadolinium d’un épendymome supratentoriel, caractérisé par une lésion bien délimitée du lobe frontal gauche avec prise de contraste hétérogène et des zones de nécrose.
Épendymome supratentoriel avec fusion YAP1-MAMLD1 :
Chez les patients touchés par ce sous-type, des chercheurs ont mis en évidence une fusion des gènes YAP1 et MAMLD1. En effet, les génomes tumoraux ont été étudiés et ont révélé des altérations du locus YAP1 à 11q22.1–11q21.2 (7/14), du locus MAMLD1 (Xp28) (10/14) et des pertes du bras chromosomique 22q (5/14). La protéine YAP1 est notamment connue pour son implication dans les voies de signalisations cellulaires Wnt et de la β-caténine, permettant une prolifération plus active des cellules de tumeurs gliales. De plus, il a été démontré que cette dernière favorise la résistance aux thérapies ciblées BRAF et MEK dans les cellules de mélanome et de cancer du poumon. Cependant à l'heure actuelle, peu de données cliniques, histopathologiques et génétiques sont disponibles sur les épendymomes avec fusions YAP1‐MAMLD1, et bien qu’aucune étude de population n’ait abordé la question jusqu'à présent, ce sous-type moléculaire semble rare [7].
Épendymome supratentoriel avec fusion ZFTA-RELA :
La fusion oncogénique entre ZFTA (Zinc Finger Translocation Associated) et RELA (REL-associated protein) représente plus de 60% des épendymomes supratentoriels. RELA est un facteur de transcription qui est impliqué dans la voie NF-κB, permettant de réguler différents mécanismes cellulaires tels que l’inflammation, la chimiotaxie et le métabolisme. D’un autre côté, les fonctions de la protéine ZFTA ne sont pas encore caractérisées [8]. Cependant, la fusion avec le gène RELA entraîne une translocation nucléaire constitutive, qui une fois dans le noyau, cible les gènes impliqués dans la voie NF-κB et entraîne une hyper-activation [9].
Épendymome supratentoriel avec délétion de CDKN2A :
Le gène CDKN2A (Cyclin Dependent Kinase inhibitor 2A), localisé sur le chromosome 9, code pour deux protéines anti-tumorales produites après un mécanisme d’épissage alternatif : p16ink4A et p14arf [10]. La délétion homozygote de CDKN2A peut entraîner une croissance cellulaire incontrôlée en l’absence de mécanisme suppresseur de tumeur efficace médié par CDKN2A. Toutefois, bien que le nombre de patients porteur de cette délétion soit faible, elle est associée à un pronostic plus sombre [3].
Épendymomes de la fosse postérieure
[modifier | modifier le code]Les épendymomes localisés dans la fosse postérieure (PF) sont des tumeurs radiologiquement homogènes et bien délimitées, avec des remaniements hémorragiques et d'éventuelles taches de calcification montrant une prise de contraste variable due à la nécrose et à la formation de kystes. Ces tumeurs peuvent être localisées à l'intérieur du quatrième ventricule avec une expansion latérale possible à travers les foramens de Luschka ou le foramen de Magendie. Les épendymomes PF peuvent être divisés en deux sous-groupes en fonction de leur profil épigénétique : les PF de groupe A (PFA) et les PF de groupe B (PFB) [11].
Ependymome PFA :
Contrairement aux épendymomes supratentoriels, les tumeurs PFA ne présentent pas de mutation somatique récurrente. Cependant, leur profil épigénétique est caractéristique, et peut comprendre une hyperméthylation des îlots CpG ou une perte de la triméthylation de l' histone : H3 en position K27. En effet, cette perte de triméthylation est fréquente chez les nourrissons de moins de 18 mois et caractéristique des PFA [7].
Ependymome PFB :
Quant à elles, les tumeurs PF-B se distinguent par un génome relativement hypométhylé, la conservation de la triméthylation H3K27 ainsi qu’un profil chromosomique polyploïde. Les PFB présentent un meilleur pronostic que les PFA, permettant une chirurgie puis une chimiothérapie adjuvante [11].
Ependymome myxopapillaire
[modifier | modifier le code]L’EP myxopapillaire est l’un des EP avec le meilleur pronostic, avec une survie à 10 ans supérieur à 90%. Situé dans la moelle épinière, l’EP myxopapillaire possède des caractéristiques morphologiques et immunohistochimiques typiques permettant un diagnostic aisé dans la plupart des cas. En cas de difficulté diagnostique, leur profil de méthylation caractéristique permet également de faciliter leur diagnostic.
Épendymomes spinaux
[modifier | modifier le code]Ils sont définis d’après l’OMS comme des tumeurs de localisation spinale bien délimitées, présentant sur le plan histologique des rosettes et sont constitués de petites cellules avec un noyau arrondi, entourées d’une matrice fibrillaire avec une faible activité mitotique. Ils ne présentent pas d’amplification de MYCN. Ils représentent 20,6% des tumeurs primitives spinales des enfants et adolescents [12] et 17,6% des jeunes adultes âgés de plus de 20 ans. Une importante proportion de cas se développent dans un contexte de neurofibromatose de type 2 [13]. Leur aspect est le plus souvent celui de cellules gliales avec un noyau rond ou ovalaire, entourées d’une matrice fibrillaire, avec fréquentes formations de pseudorosettes. L’immunophénotype est caractéristique, avec une expression de la GFAP, de la protéine S100 et de la vimentine par les cellules tumorales, avec une expression focale de l’EMA. En cas de morphologie difficile à aborder, une analyse de leur profil de méthylation qui est caractéristique permet d’orienter le diagnostic.
Traitement
[modifier | modifier le code]Les PFA sont les épendymomes intracrâniens les plus complexes à soigner, de par leur localisation latérale prédominante mais aussi de par leur pronostic péjoratif. La faible accessibilité chirurgicale rend par ailleurs l'exérèse complète difficile, augmentant le risque de récidive. Au contraire, les PFB sont associés à un pronostic plus favorable car ils sont plus facilement opérables et présentent une biologie moins agressive. En effet, plusieurs analyses de cohortes de patients ont montré que des patients PFA ayant reçu une intervention chirurgicale et une radiothérapie récidivent plus que les patients PFB, dont aucun patient n’a récidivé après résection totale de la tumeur sans radiothérapie.
La radiothérapie s’ajoute à la résection chirurgicale en cas de tumeur non disséminée, dans le but de contrôler la récidive locale. Comme pour la chirurgie, les PFA restent également difficiles à traiter de par leur proximité avec des structures neurologiques importantes.
Concernant les autres types tumoraux (EP avec fusion ZFTA-RELA ou YAP1-MAMLD1), la même cohorte n'a pu mettre en évidence de preuve que la résection totale améliorerait la survie sans progression. Enfin, les EP myxopapillaires peuvent être traités par un acte chirurgical, avec de la radiothérapie adjuvante en cas de résection incomplète pour améliorer le pronostic [14].
La chimiothérapie est généralement réservée aux cas d'épendymome avancés ou récurrents qui ne peuvent pas être réséqués ou irradiés de manière efficace, mais n’a actuellement pas démontré d’avantage sur la survie des patients [15],[4].
Illustrations
[modifier | modifier le code]-
 Radiographie d'un épendymome Archives militaires médicales
Radiographie d'un épendymome Archives militaires médicales -
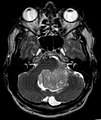 Épendymome du ventricule 4
Épendymome du ventricule 4 -
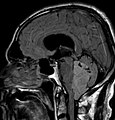 Épendymome du ventricule 4
Épendymome du ventricule 4 -
 Épendymome du ventricule 4. (à droite avec contraste augmenté)
Épendymome du ventricule 4. (à droite avec contraste augmenté)




Notes et références
[modifier | modifier le code]- Canadian Cancer Society / Société canadienne du cancer, « Épendymome », sur Société canadienne du cancer (consulté le ).
- (en) Quinn T Ostrom, Nirav Patil, Gino Cioffi et Kristin Waite, « CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017 », Neuro-Oncology, vol. 22, no Supplement_1, , iv1–iv96 (ISSN 1522-8517 et 1523-5866, DOI 10.1093/neuonc/noaa200, lire en ligne, consulté le )
- (en) Thomas Larrew, Brian Fabian Saway, Stephen R. Lowe et Adriana Olar, « Molecular Classification and Therapeutic Targets in Ependymoma », Cancers, vol. 13, no 24, , p. 6218 (ISSN 2072-6694, DOI 10.3390/cancers13246218, lire en ligne, consulté le )
- (en) Roberta Rudà, Guido Reifenberger, Didier Frappaz et Stefan M Pfister, « EANO guidelines for the diagnosis and treatment of ependymal tumors », Neuro-Oncology, vol. 20, no 4, , p. 445–456 (ISSN 1522-8517 et 1523-5866, DOI 10.1093/neuonc/nox166, lire en ligne, consulté le )
- (en) David N Louis, Arie Perry, Pieter Wesseling et Daniel J Brat, « The 2021 WHO Classification of Tumors of the Central Nervous System: a summary », Neuro-Oncology, vol. 23, no 8, , p. 1231–1251 (ISSN 1522-8517 et 1523-5866, DOI 10.1093/neuonc/noab106, lire en ligne, consulté le )
- « Épendymome : symptômes de cette tumeur du système nerveux », sur passeportsante.net, (consulté le ).
- (en) Felipe Andreiuolo, Pascale Varlet, Arnault Tauziède‐Espariat et Stephanie T. Jünger, « Childhood supratentorial ependymomas with YAP1‐MAMLD1 fusion: an entity with characteristic clinical, radiological, cytogenetic and histopathological features », Brain Pathology, vol. 29, no 2, , p. 205–216 (ISSN 1015-6305 et 1750-3639, DOI 10.1111/bpa.12659, lire en ligne, consulté le )
- (en) Amir Arabzade, Yanhua Zhao, Srinidhi Varadharajan et Hsiao-Chi Chen, « ZFTA–RELA Dictates Oncogenic Transcriptional Programs to Drive Aggressive Supratentorial Ependymoma », Cancer Discovery, vol. 11, no 9, , p. 2200–2215 (ISSN 2159-8274 et 2159-8290, DOI 10.1158/2159-8290.CD-20-1066, lire en ligne, consulté le )
- (en) Vicente Herranz-Pérez, Jin Nakatani, Masaki Ishii et Toshiaki Katada, « Ependymoma associated protein Zfta is expressed in immature ependymal cells but is not essential for ependymal development in mice », Scientific Reports, vol. 12, no 1, (ISSN 2045-2322, DOI 10.1038/s41598-022-05526-y, lire en ligne, consulté le )
- (en) Alexander Yuile, Laveniya Satgunaseelan, Joe Q. Wei et Michael Rodriguez, « CDKN2A/B Homozygous Deletions in Astrocytomas: A Literature Review », Current Issues in Molecular Biology, vol. 45, no 7, , p. 5276–5292 (ISSN 1467-3045, DOI 10.3390/cimb45070335, lire en ligne, consulté le )
- (en) Johannes Nowak, Stephanie Theresa Jünger, Henner Huflage et Carolin Seidel, « MRI Phenotype of RELA-fused Pediatric Supratentorial Ependymoma », Clinical Neuroradiology, vol. 29, no 4, , p. 595–604 (ISSN 1869-1439 et 1869-1447, DOI 10.1007/s00062-018-0704-2, lire en ligne, consulté le )
- (en) Martin Benesch, Didier Frappaz et Maura Massimino, « Spinal cord ependymomas in children and adolescents », Child's Nervous System, vol. 28, no 12, , p. 2017–2028 (ISSN 0256-7040 et 1433-0350, DOI 10.1007/s00381-012-1908-4, lire en ligne, consulté le )
- (en) Ashok R Asthagiri, Dilys M Parry, John A Butman et H Jeffrey Kim, « Neurofibromatosis type 2 », The Lancet, vol. 373, no 9679, , p. 1974–1986 (DOI 10.1016/S0140-6736(09)60259-2, lire en ligne, consulté le )
- (en) Stephanie Theresa Jünger, Beate Timmermann et Torsten Pietsch, « Pediatric ependymoma: an overview of a complex disease », Child's Nervous System, vol. 37, no 8, , p. 2451–2463 (ISSN 0256-7040 et 1433-0350, DOI 10.1007/s00381-021-05207-7, lire en ligne, consulté le )
- (en) Kristian W. Pajtler, Stephen C. Mack, Vijay Ramaswamy et Christian A. Smith, « The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants », Acta Neuropathologica, vol. 133, no 1, , p. 5–12 (ISSN 0001-6322 et 1432-0533, DOI 10.1007/s00401-016-1643-0, lire en ligne, consulté le )