The evolutionary history of 2,658 cancers
- PMID: 32025013
- PMCID: PMC7054212
- DOI: 10.1038/s41586-019-1907-7
The evolutionary history of 2,658 cancers
Erratum in
-
Author Correction: The evolutionary history of 2,658 cancers.Nature. 2023 Feb;614(7948):E42. doi: 10.1038/s41586-022-05601-4. Nature. 2023. PMID: 36697833 Free PMC article. No abstract available.
Abstract
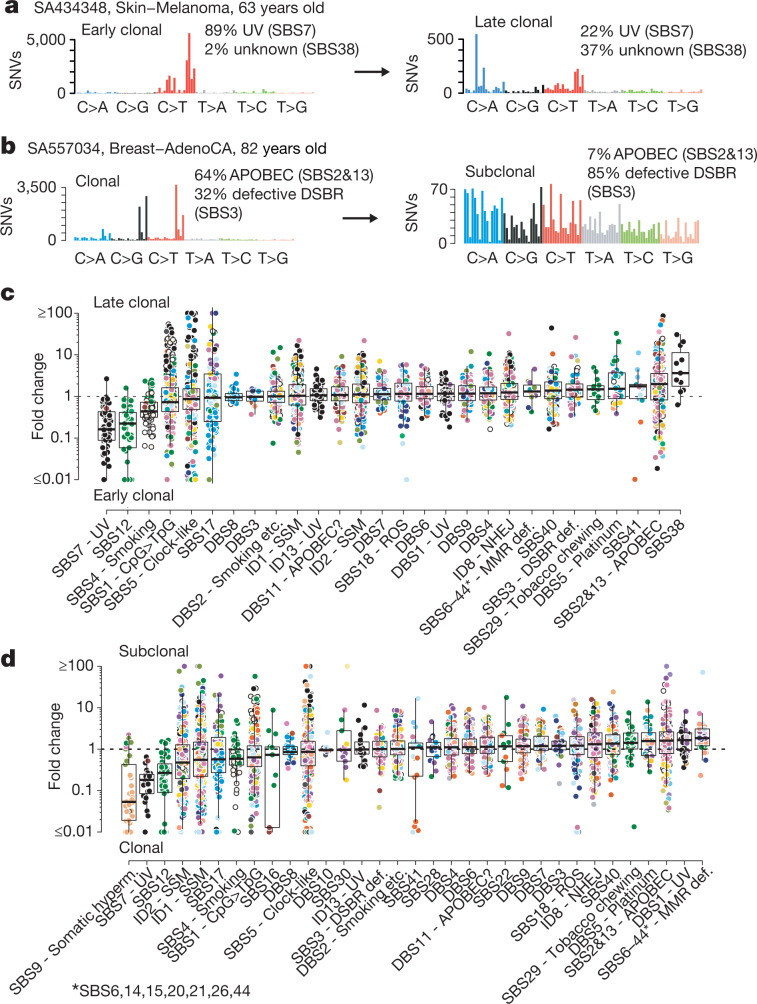
Cancer develops through a process of somatic evolution1,2. Sequencing data from a single biopsy represent a snapshot of this process that can reveal the timing of specific genomic aberrations and the changing influence of mutational processes3. Here, by whole-genome sequencing analysis of 2,658 cancers as part of the Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium of the International Cancer Genome Consortium (ICGC) and The Cancer Genome Atlas (TCGA)4, we reconstruct the life history and evolution of mutational processes and driver mutation sequences of 38 types of cancer. Early oncogenesis is characterized by mutations in a constrained set of driver genes, and specific copy number gains, such as trisomy 7 in glioblastoma and isochromosome 17q in medulloblastoma. The mutational spectrum changes significantly throughout tumour evolution in 40% of samples. A nearly fourfold diversification of driver genes and increased genomic instability are features of later stages. Copy number alterations often occur in mitotic crises, and lead to simultaneous gains of chromosomal segments. Timing analyses suggest that driver mutations often precede diagnosis by many years, if not decades. Together, these results determine the evolutionary trajectories of cancer, and highlight opportunities for early cancer detection.
Conflict of interest statement
R.B. owns equity in Ampressa Therapeutics. G.G. receives research funds from IBM and Pharmacyclics and is an inventor on patent applications related to MuTect, ABSOLUTE, MutSig, MSMuTect and POLYSOLVER. I.L. is a consultant for PACT Pharma. B.J.R. is a consultant at and has ownership interest (including stock and patents) in Medley Genomics. All other authors declare no competing interests.
Figures














Comment in
-
Global genomics project unravels cancer's complexity at unprecedented scale.Nature. 2020 Feb;578(7793):39-40. doi: 10.1038/d41586-020-00213-2. Nature. 2020. PMID: 32025004 No abstract available.
Similar articles
-
Pan-cancer analysis of whole genomes.Nature. 2020 Feb;578(7793):82-93. doi: 10.1038/s41586-020-1969-6. Epub 2020 Feb 5. Nature. 2020. PMID: 32025007 Free PMC article.
-
Patterns of somatic structural variation in human cancer genomes.Nature. 2020 Feb;578(7793):112-121. doi: 10.1038/s41586-019-1913-9. Epub 2020 Feb 5. Nature. 2020. PMID: 32025012 Free PMC article.
-
Whole-genome sequencing of phenotypically distinct inflammatory breast cancers reveals similar genomic alterations to non-inflammatory breast cancers.Genome Med. 2021 Apr 26;13(1):70. doi: 10.1186/s13073-021-00879-x. Genome Med. 2021. PMID: 33902690 Free PMC article.
-
Decoding human cancer with whole genome sequencing: a review of PCAWG Project studies published in February 2020.Cancer Metastasis Rev. 2021 Sep;40(3):909-924. doi: 10.1007/s10555-021-09969-z. Epub 2021 Jun 7. Cancer Metastasis Rev. 2021. PMID: 34097189 Free PMC article. Review.
-
Cancer heterogeneity: converting a limitation into a source of biologic information.J Transl Med. 2017 Sep 8;15(1):190. doi: 10.1186/s12967-017-1290-9. J Transl Med. 2017. PMID: 28886708 Free PMC article. Review.
Cited by
-
Molecular profiling reveals novel therapeutic targets and clonal evolution in ovarian clear cell carcinoma.BMC Cancer. 2024 Nov 14;24(1):1403. doi: 10.1186/s12885-024-13125-5. BMC Cancer. 2024. PMID: 39543535 Free PMC article.
-
Deletions Rate-Limit Breast and Ovarian Cancer Initiation.bioRxiv [Preprint]. 2024 Oct 21:2024.10.17.618945. doi: 10.1101/2024.10.17.618945. bioRxiv. 2024. PMID: 39484366 Free PMC article. Preprint.
-
Polyclonal-to-monoclonal transition in colorectal precancerous evolution.Nature. 2024 Dec;636(8041):233-240. doi: 10.1038/s41586-024-08133-1. Epub 2024 Oct 30. Nature. 2024. PMID: 39478225
-
Temporal recording of mammalian development and precancer.Nature. 2024 Oct;634(8036):1187-1195. doi: 10.1038/s41586-024-07954-4. Epub 2024 Oct 30. Nature. 2024. PMID: 39478207 Free PMC article.
-
Polyclonality overcomes fitness barriers in Apc-driven tumorigenesis.Nature. 2024 Oct;634(8036):1196-1203. doi: 10.1038/s41586-024-08053-0. Epub 2024 Oct 30. Nature. 2024. PMID: 39478206 Free PMC article.
References
-
- Cairns, J. Mutation selection and the natural history of cancer. Nature255, 197–200 (1975). - PubMed
-
- Martincorena, I. & Campbell, P. J. Somatic mutation in cancer and normal cells. Science349, 1483–1489 (2015). - PubMed
-
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature10.1038/s41586-020-1969-6 (2020).
-
- Moore, L. et al. The mutational landscape of normal human endometrial epithelium. Preprint at bioRxiv 10.1101/505685 (2018).
Publication types
MeSH terms
Grants and funding
- 27176/CRUK_/Cancer Research UK/United Kingdom
- U24 CA143799/CA/NCI NIH HHS/United States
- MR/L016311/MRC_/Medical Research Council/United Kingdom
- 28290/CRUK_/Cancer Research UK/United Kingdom
- P01 CA206978/CA/NCI NIH HHS/United States
- 1U24CA143799/CA/NCI NIH HHS/United States
- R01 CA183793/CA/NCI NIH HHS/United States
- U24 CA210957/CA/NCI NIH HHS/United States
- R01 CA132897/CA/NCI NIH HHS/United States
- 15973/CRUK_/Cancer Research UK/United Kingdom
- U24 CA211000/CA/NCI NIH HHS/United States
- P30 CA016672/CA/NCI NIH HHS/United States
- MR/L016311/1/MRC_/Medical Research Council/United Kingdom
- FC001202/ARC_/Arthritis Research UK/United Kingdom
- 19274/CRUK_/Cancer Research UK/United Kingdom
- MH086633/MH/NIMH NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- U24 CA210999/CA/NCI NIH HHS/United States
- GM108308/NH/NIH HHS/United States
- FC001202/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources